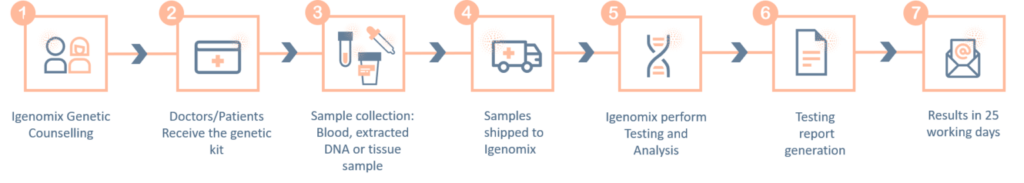
Polycystic Kidney Disease Precision Panel
Polycystic Kidney Disease (PKD) is an inherited multisystemic and progressive disorder characterized by cyst formation and enlargement of the kidneys and other organs. Cysts are noncancerous round sacs containing fluid.